

TNF alpha/TNFA/TNFSF2免疫信号通路及重组蛋白研究分析
- 2026-05-22
- 119
肿瘤坏死因子α(Tumor Necrosis Factor-α,TNF-α),编码基因为TNFA(又称TNFSF2),是肿瘤坏死因子超家族(TNFSF)的核心成员,作为多功能促炎细胞因子,在先天免疫应答、炎症调控、细胞命运决定及免疫稳态维持中发挥核心作用。
TNF-α的生理功能是依靠自身介导的信号传导通路实现的,该因子与细胞膜表面特异性受体结合后启动下游信号级联反应并调控细胞增殖凋亡、炎症介质分泌释放等一系列生理过程。TNF-α 参与机体正常免疫防御同时与自身免疫疾病、恶性肿瘤、脓毒症等多种病症存在密切关联。深入研究TNF-α信号传导通路的分子作用机制,能够明确其生理与病理功能并为靶向干预方案及重组蛋白类药物开发提供核心依据。
一、TNF-α信号转导通路
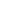
TNF-α以同源三聚体形式存在,分为膜结合型(mTNF-α)与可溶性(sTNF-α)两种活性形式,后者由前者经金属蛋白酶剪切释放,二者分别介导局部与远距离信号传导。其信号转导依赖细胞膜上的TNFR1和TNFR2两种特异性受体,二者同属TNFR超家族,表达分布、信号机制及功能差异显著,形成核心信号分支并交叉调控,共同决定细胞应答结果。
TNFR1广泛分布于机体各类组织细胞并带有死亡结构域可调控炎症反应细胞存活细胞凋亡与坏死性凋亡相关信号通路;TNFR2主要分布在免疫细胞和血管内皮细胞不含死亡结构域主要介导免疫调节细胞存活和组织修复相关信号传导。两类受体结合不同衔接蛋白激活下游信号TNFR2可竞争性抑制TNFR1的促炎与凋亡信号共同形成复杂的信号调控网络,受体系统功能异常参与多种疾病发生发展过程,为重组蛋白药物的靶点开发提供重要理论依据。
![图1 肿瘤坏死因子-α(TNF):TNF受体(TNFR)介导的细胞信号(源于[1]).png](/uploads/images/20260522/1779435834873713.png)
图1 肿瘤坏死因子-α(TNF)/TNF受体(TNFR)介导的细胞信号(源于[1])
二、免疫缺陷病毒1型(HIV-1)蛋白靶点
免疫缺陷病毒1型(HIV-1)的复制周期依赖其编码的多种结构蛋白和非结构蛋白,这些蛋白是抗病毒药物研发及重组蛋白研究的核心靶点。其中HIV-1编码的Vpr、Tat、Nef及gp120等蛋白,会通过干扰该通路,异常激活NF-κB等转录因子,增强感染细胞中HIV转录,加速病毒复制。
病毒结构蛋白里,env基因翻译合成的gp120与gp41属于重要作用位点,其中gp120可识别并结合宿主细胞CD4受体与共受体CCR5/CXCR4,完成病毒囊膜与宿主细胞质膜的融合过程;gag基因表达的p24核衣壳蛋白则参与病毒粒子的装配与后期成熟。此外,pol基因编码的逆转录酶、蛋白酶、整合酶是病毒非结构蛋白中的核心作用位点,三类酶分别负责病毒RNA逆转录为DNA、病毒前体蛋白的切割加工以及病毒DNA整合进入宿主基因组。这些蛋白是临床抗病毒药物的主要作用位点,也为重组蛋白疫苗及相关抑制剂开发提供重要研究基础。
![图2 人类免疫缺陷病毒1型(HIV-1)蛋白靶向TNF:TNFR信号通路(源于[1]).png](/uploads/images/20260522/1779435881552079.png)
图2 人类免疫缺陷病毒1型(HIV-1)蛋白靶向TNF/TNFR信号通路(源于[1])
三、免疫调控与炎症应答
TNF/TNFR 信号轴承担免疫调节与炎症反应的核心功能,可精准调控固有免疫与适应性免疫细胞活性,维持机体免疫状态稳定和炎症水平均衡。该信号轴能够激活巨噬细胞、树突状细胞等固有免疫细胞,促进促炎介质分泌,启动炎症反应以清除入侵病原体;同时调控T/B淋巴细胞增殖分化过程,提升适应性免疫应答效率,从而构建完善的免疫防御系统。
当HIV-1蛋白异常作用于TNF/TNFR信号轴时,会破坏免疫调节与炎症反应的稳定状态,持续激活慢性炎症并伴随大量促炎介质释放,造成组织损伤。同时也会抑制免疫细胞正常功能,引发免疫耗竭,减弱机体对HIV-1的防御能力,形成炎症紊乱与免疫缺陷相互加重的恶性循环,这一过程是HIV-1感染后机体免疫功能失调的关键机制。
四、TNF/TNFR通路分子调控机制
TNF/TNFR通路的分子调控依赖受体介导的信号复合物组装与逐级信号激活,TNFR1通过募集TRADD、TRAF2等衔接蛋白,调控NF-κB、MAPK等下游通路,实现促炎反应细胞存活与细胞凋亡的动态转换。TNFR2通过激活 PI3K/Akt通路,参与免疫调节与组织修复,两种受体共同维持通路正常功能。
HIV-1蛋白通过特异性分子相互作用来干扰信号调控过程。该分子层面的异常相互作用明确了HIV-1的致病机制,同时为靶向TNF/TNFR通路的重组蛋白药物开发提供了精准分子靶点。其中Vpr、Tat等蛋白可直接结合通路关键信号分子来异常激活NF-κB通路;gp120通过改变TNFR表达水平来增强TNF-α介导的信号传导,进而促进HIV-1的转录与复制。
五、疾病关联及靶向药物研发进展
TNF/TNFR通路异常激活或被干扰,与多种疾病的发生发展密切相关。除HIV-1感染引发的免疫缺陷及慢性炎症外,通路过度激活可介导类风湿关节炎、强直性脊柱炎等自身免疫病,还与肿瘤进展、脓毒症等密切相关;通路抑制则会导致免疫功能低下,增加病原体感染风险。
目前药物研发主要聚焦于靶向TNF/TNFR通路的抑制剂及重组蛋白,其中重组TNF-α拮抗剂、重组TNFR融合蛋白已应用于自身免疫病治疗;针对HIV-1感染,靶向通路关键分子及HIV-1蛋白的重组蛋白药物正处于研发阶段,通过阻断HIV-1对通路的干扰,抑制病毒复制,为HIV-1防治及相关并发症干预提供了新方向。
卡梅德生物(KMD Bioscience)依托完善的重组蛋白研发平台,构建了覆盖原核、酵母、哺乳及无细胞表达系统的全流程技术体系,可高效表达并精准修饰重组蛋白,提供高纯度、高活性的抗原产品。我们提供从抗原设计、重组蛋白制备到药物候选物筛选的一体化解决方案,助力靶向免疫治疗项目新突破,加速创新药物转化。
TNF alpha/TNFA/TNFSF2靶点相关产品包括
Protein | ||
Catalog Number | Product Name | Expression System |
KMPH4570 | E. coli | |
KMH1819 | E. coli | |
KMH1820 | HEK293/E.coli | |
KMP1885 | HEK293 | |
KMP2130 | / | |
KMP2133 | / | |
Antibody | ||
Catalog Number | Product Name | Application |
YR1005 | FCM, IP, ELISA, Neut, FuncS | |
YR1094 | IF, IP, Neut, FuncS, ELISA, FCM, WB | |
YR1111 | IP, IF, FuncS, FCM, Neut, ELISA, WB | |
YR1137 | / | |
YR1205 | FuncS, IF, Neut, ELISA, FCM, IP, ICC | |
YR1259 | / | |
YR1289 | WB, FCM, IP, ELISA, Neut, FuncS, IF | |
YR1349 | / | |
YR1612 | / | |
YR1625 | / | |
PA7253 | WB, IF | |
PA7310 | WB, IF, IP | |
PA11024 | WB | |
PA11484 | WB | |
PA12786 | WB | |
PA14796
| WB, IHC | |
PA16275 | WB | |
MA610 | WB, IHC | |
YR1068 | Neut, ELISA, IF, IP, FuncS, FCM, ICC |
Q 1:TNF-α的膜结合型(mTNF-α)与可溶性(sTNF-α)在功能上有何本质区别?
A:TNF-α以同源三聚体形式存在,分为膜结合型(mTNF-α)和可溶性(sTNF-α)两种活性形式,在合成、释放及功能上存在显著差异。mTNF-α是TNF-α的初始前体形式,通过跨膜结构域锚定在细胞膜表面,主要表达于活化的巨噬细胞、T细胞和自然杀伤细胞等免疫细胞上。它介导局部的细胞间接触依赖信号,尤其对邻近靶细胞产生作用,并能够反向信号传导调节自身细胞功能。sTNF-α则由mTNF-α经金属蛋白酶剪切释放进入血液循环,具有远距离内分泌作用,是全身性炎症反应的主要介质。
在功能分工上,mTNF-α更倾向于激活TNFR2,参与免疫调节、组织修复和淋巴细胞稳态;而sTNF-α主要结合TNFR1,驱动强烈的促炎反应和细胞凋亡。由于sTNF-α半衰期短、扩散能力强,其异常升高与脓毒症、类风湿关节炎等全身性炎症疾病密切相关;mTNF-α则更多参与局部免疫突触形成和慢性炎症微环境维持。
Q 2:TNFR1与TNFR2在信号转导机制和功能上有哪些核心差异?
A:TNFR1和TNFR2虽同属TNFR超家族,但结构、表达分布及下游信号路径截然不同。TNFR1广泛表达于几乎所有有核细胞,其胞内区含有一个死亡结构域(DD),能够招募TRADD、FADD和RIPK1等衔接蛋白,从而启动三条主要信号通路:NF-κB通路、凋亡通路以及坏死性凋亡通路。因此,TNFR1是一个“双刃剑”:在正常生理下维持细胞存活与炎症防御,在过度激活或caspase-8被抑制时则诱导细胞死亡。
相比之下,TNFR2主要表达于免疫细胞和内皮细胞,其胞内区无死亡结构域,而是通过TRAF2、cIAP1/2等蛋白激活NF-κB和PI3K/Akt通路,主要促进细胞增殖、存活、组织修复及免疫调节。TNFR2信号能够增强调节性T细胞的抑制功能,并促进CD8+ T细胞的记忆形成。值得注意的是,TNFR2可竞争性结合TNF-α,尤其是与mTNF-α亲和力更高,从而限制TNFR1的促凋亡信号。这种受体间的交叉调控形成了复杂的信号网络:在HIV-1感染中,病毒蛋白常通过上调TNFR2表达来干扰免疫稳态,促进免疫耗竭。
Q 3:HIV-1病毒蛋白如何具体干扰TNF/TNFR通路以增强自身复制?
A:HIV-1编码的多种蛋白通过直接或间接方式劫持TNF/TNFR信号轴,异常激活下游转录因子NF-κB,从而促进病毒基因转录。具体机制包括:Vpr蛋白可与宿主细胞内的TRADD、GR以及p65(RelA)相互作用,增强TNFR1介导的NF-κB核转位,同时抑制糖皮质激素受体介导的抗炎效应;Tat蛋白则作为强效转录激活因子,不仅能与Cyclin T1结合促进RNA聚合酶II延伸,还能直接激活IKK复合物,导致IκBα磷酸化降解,使NF-κB持续入核。
此外,gp120通过与宿主CD4受体及共受体CCR5/CXCR4结合,激活下游PI3K/Akt和MAPK通路,间接上调TNFR1和TNFR2的表面表达水平,使细胞对TNF-α信号高度敏感,进一步放大NF-κB驱动的HIV-1长末端重复序列(LTR)转录活性。Nef蛋白则通过下调MHC-I和CD4逃避宿主免疫,同时激活PAK2激酶促进NF-κB入核。这些病毒蛋白的协同作用导致慢性炎症微环境中HIV-1复制效率显著提升,同时诱导T细胞凋亡和免疫耗竭,形成“炎症-病毒复制-免疫损伤”的恶性循环。
Q 4:TNF/TNFR信号轴在维持免疫稳态与炎症应答中扮演何种双重角色?
A:在生理状态下,TNF/TNFR信号轴是固有免疫和适应性免疫的关键调节枢纽。一方面,当病原体入侵时,局部释放的TNF-α激活巨噬细胞和树突状细胞,促进IL-1、IL-6、趋化因子等促炎介质分泌,增强吞噬作用和抗原提呈能力,从而快速清除病原体。同时,TNF-α通过TNFR2信号促进调节性T细胞(Treg)的扩增和功能维持,防止过度炎症损伤组织——这是其“免疫调节”的一面。
另一方面,TNF-α也参与适应性免疫的精细调控,通过促进T细胞向Th1分化、B细胞抗体类别转换以及生发中心形成,增强免疫记忆。然而,当HIV-1蛋白持续异常激活该通路时,TNF-α的促炎作用占主导地位,导致慢性炎症状态,表现为血液中IL-6、CRP、TNF-α本身持续升高。这种慢性炎症不仅造成CD4+ T细胞过度活化、易发生activation-induced cell death(AICD),还诱导免疫检查点分子高表达,驱动T细胞耗竭。同时,TNF-α介导的纤维化和淋巴组织破坏进一步削弱免疫重建能力。因此,TNF/TNFR轴在“防御”与“损伤”之间的平衡一旦被HIV-1打破,便成为免疫缺陷与炎症紊乱相互加重的关键驱动因素。
Q 5:TNFR1和TNFR2下游的主要信号通路如何实现促炎、存活与凋亡的动态转换?
A:TNFR1的胞内死亡结构域在配体结合后迅速募集TRADD,后者作为平台蛋白进一步招募TRAF2、RIPK1和cIAP1/2形成复合物I。该复合物通过激活TAK1和IKK复合物,导致IκBα降解,使NF-κB二聚体(p50/p65)入核启动抗凋亡基因和促炎基因转录。同时,MAPK通路被激活,调节细胞增殖和炎症反应。如果复合物I信号不足或cIAP1/2活性下降,RIPK1会从复合物I解离并与FADD、caspase-8形成复合物IIa,启动外源性凋亡。当caspase-8被病毒蛋白或药物抑制时,RIPK1/RIPK3相互作用形成坏死小体,通过MLKL介导坏死性凋亡。
TNFR2是通过TRAF2招募cIAP1/2直接激活NF-κB和PI3K/Akt通路。PI3K/Akt信号可抑制GSK-3β、激活mTOR,促进细胞存活、蛋白质合成和代谢重编程。此外,TNFR2诱导的NF-κB信号更倾向于表达抗凋亡蛋白和免疫调节分子,而不诱导强烈的炎症因子。因此,TNFR1偏向炎症和死亡诱导,TNFR2偏向保护和修复,两种受体的动态平衡决定了细胞命运。HIV-1蛋白通过上调TNFR2或干扰复合物I的组装,可人为地转向慢性炎症或加速细胞死亡。
- 联系我们
- 电话:400-621-6806
- 手机:13116038708(微信)
- 邮箱:info@kmdbioscience.com
- 地址:天津市武清开发区和畅路6号10号厂房108室
-

销售微信
-

官方公众号
-
 400-621-6806
400-621-6806 -
 3599163699
3599163699 -
 周一至周五 9:00-18:00
周一至周五 9:00-18:00 -
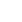
-
 0
0
-
